Protective and Pathogenic Th17 cells
A novel work from the Kuchroo’s lab contributes considerably to our understanding of the various subsets of IL-17-producing cells, with delimitation of non-pathogenic from pathogenic Th17 cells.
This is very important because even though there is increasing evidence that IL-17-producing Th17 cells are not always detrimental to host, there is still a long way to go to clearly characterize differences in the requirements for the induction and the phenotype of nonpathogenic and pathogenic Th17 cells.
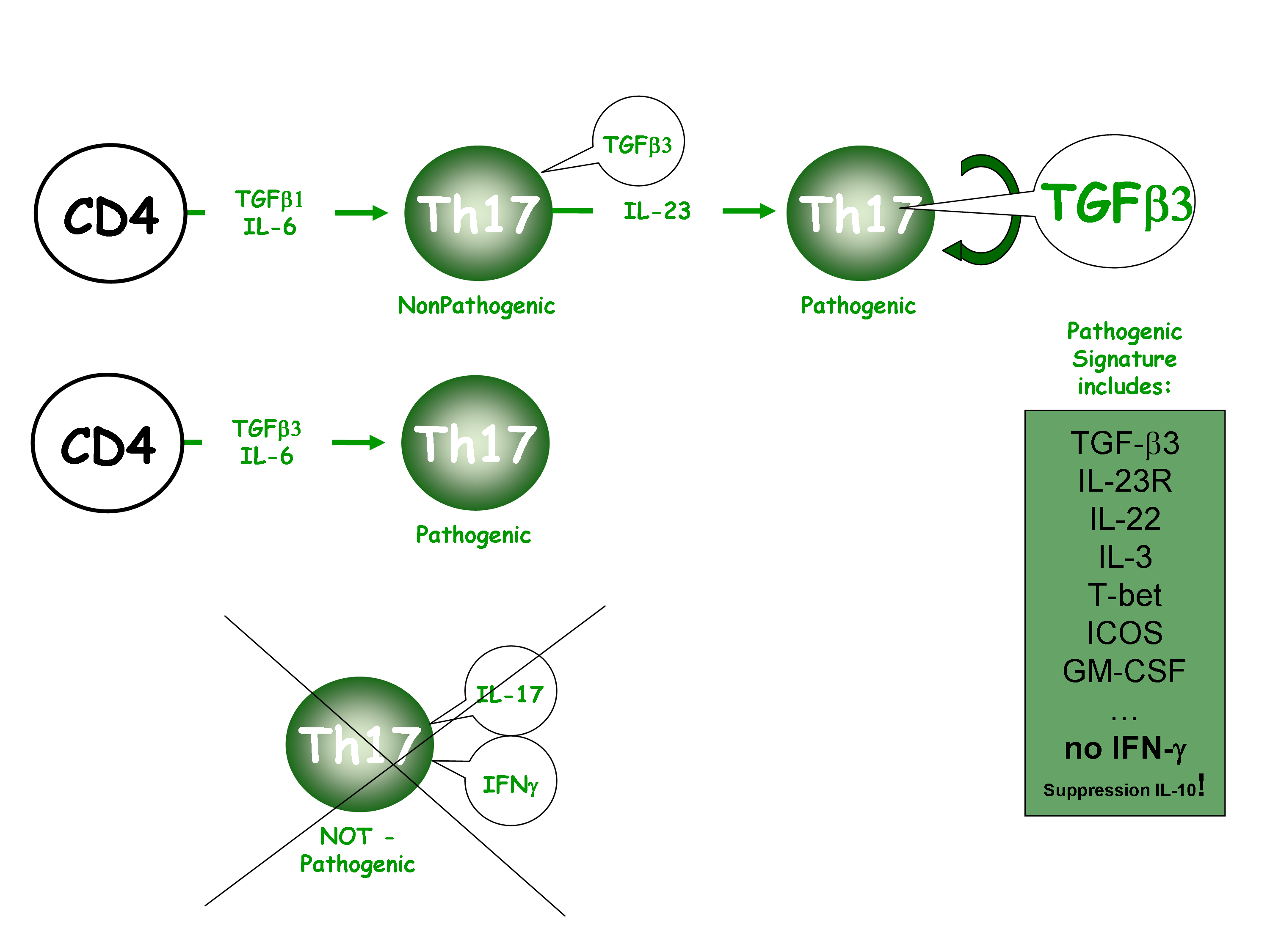
The present study sheds new light on why IL-23 is critical to confert autoimmune properties to Th17 cells. It shows that the role of the pathogenic cytokine IL-23 is to induce IL-17-producing cells to produce TGF-β3. Importantly, Th17 cells induced with TGF-β1 + IL-6 + IL-23 triggered severe EAE disease, whereas Th17 cells induced with TGF-β1 + IL-6 resulted on mild or no disease. In this context, TGF-β3 was identified as the pathogenic molecule induced through IL-23 signaling.
Then, they compared TGF-β1 + IL-6 or TGF-β3 + IL-6 polarized Th17 cells and found that TGF-β3-induced Th17 cells are highly pathogenic inducing EAE with a high disease incidence, severity and mortality. Surprisingly, although both TGF-β1 and TGF-β3 signal through TGF-βRII to control Th17 differentiation, they trigger different profile of downstream target genes, which leads to different outcomes.
TGF-β3-induced Th17 cells had significantly higher expression of Il23r and Il22, Cxcl3, Il3, Ccl4, Gzmb, Lrmp, Ccl5, Casp1, CFS2, Ccl3, Tbox21, ICOS, Il7r, Stat4 than did TGF-β1-induced Th17 cells.
Finally, whereas mice deficient in IFN-γ or its receptor are highly susceptible to the development of EAE, mice that lack T-bet are protected from almost all autoimmune disease. Intriguingly, they report that Tbox21-/- T cells incubated with TGF-β3 + IL-6 did not trigger EAE while WT incubated with TGF-β1 + IL-6 did.
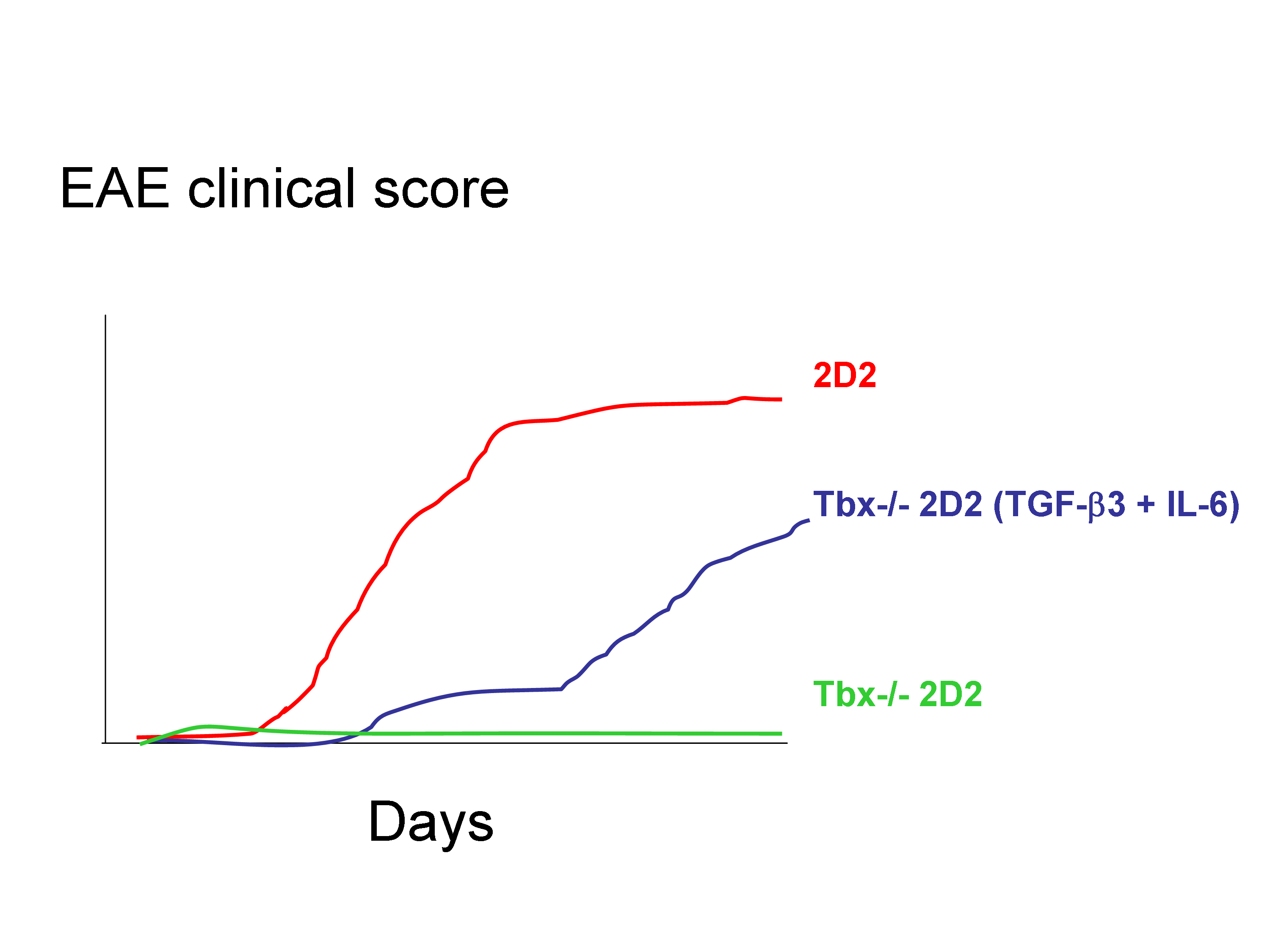
Surprisingly, Tgfb3 expression was lower in Tbox21-/- T cells than in WT T cells. Thus, one of the key results presented in this paper is that Tbx21−/− 2D2 Th17 cells induced by TGF-β3 and IL-6 were pathogenic in vivo, and thus likely independently of IFN-ɣ production. This contrast with recent data suggesting a role for IFN-ɣ in breaching the blood-brain barrier to allow the entry of IL-17-producing cells into the CNS.
These observations suggested that T-bet was part of the pathogenic Th17 signature and may have regulated the endogenous expression of TGF-β3. Loss of pathogenicity in T-bet-deficient Th17 cells was overcome by exogenous TGF-β3 and IL-6. However, it remains unclear whether T-bet is directly or indirectly required for the induction and/or maintenance of TGF-β3.